LUYOR-3109高强度紫外催化光源促销
LUYOR-3109紫外光源采用了9颗365nm大功率led,安装有二次光学透镜,输出紫外线强度高,...
2024-08-08

作者:生命科学事业部时间:2019-11-17 19:53:46浏览8338 次
慢病毒(Lentivirus)是逆转录病毒的一种。构建的siRNA / miRNA慢病毒载体,与化学合成的siRNA 和基于瞬时表达载体构建的普通 siRNA 载体相比,一方面可以扩增替代瞬时表达载体使用,另一方面,Lentivirus-siRNA克隆经过慢病毒包装系统包装后,可用于感染依靠传统转染试剂难于转染的细胞系如原代细胞、悬浮细胞和处于非分裂状态的细胞,并且在感染后可以整合到受感染细胞的基因组,进行长时间的稳定表达。
目的基因不能直接整合到大多数真核细胞,常用的手段是将目的基因包装成病毒来感染细胞,从而得到表达满足实验需求。
1、病毒的种类
病毒有很多种,常见的有慢病毒和腺病毒
1.1慢病毒
1.1.1原理
慢病毒(Lentivirus)是逆转录病毒的一种。构建的siRNA / miRNA慢病毒载体,与化学合成的siRNA 和基于瞬时表达载体构建的普通 siRNA 载体相比,一方面可以扩增替代瞬时表达载体使用,另一方面,Lentivirus-siRNA克隆经过慢病毒包装系统包装后,可用于感染依靠传统转染试剂难于转染的细胞系如原代细胞、悬浮细胞和处于非分裂状态的细胞,并且在感染后可以整合到受感染细胞的基因组,进行长时间的稳定表达。
1.1.2特点
1)直接包装成为假病毒颗粒,对分裂和非分裂细胞均有感染作用,适合 RNAi 研究和体内实验中难于转染的细胞 (比如神经元细胞、干细胞或其它原代细胞)。
2)可以通过简单方式,在短时间内获得稳定表达特定基因的多种细胞株。
3)可用于基因敲除、基因治疗和转基因动物研究。
4)无需任何转染试剂,操作简便。
5)可以根据客户需要制备多种标记。
1.1.3慢病毒包装简要流程:
1)含有目的基因的慢病毒 RNAi 干扰载体的构建和质粒纯化提取。
2)慢病毒载体,包装系统共转染病毒包装细胞293T等。
3)培养 48hrs - 72hrs 左右,收集含有病毒的上清培养液。
4)病毒的纯化和浓缩。
5)分装、- 80 ℃保存。
6)滴度测定目的基因检定,并出具检测报告。
1.2、腺病毒
1.2.1原理
腺病毒(Adenovirus,Ad)是一种无包膜的线状双链DNA病毒,其复制不依赖于宿主细胞的分裂。有近50个血清型,大多数Ad载体都是基于血清型2和5,通过转基因的方式取代E1和E3基因,降低病毒的复制能力。这些重组病毒仅在高水平表达E1和E3基因的细胞中复制,因此是一种适用于治疗的控制系统。
1.2.2特点
1)几乎可以感染所有类型的细胞
2)可以获得复制缺陷型 (E1 和 E3 缺失) 的腺病毒
3)病毒滴度高,产生病毒经过浓缩后可以达到 1012 PFU/mL,能有效的进行增殖。
4)腺病毒载体感染宿主的范围比较广,制备容易,操作简单.
5)感染细胞时,不整合到染色体中,不存在激活致癌基因或插入突变等危险,生物安全性高。
1.2.3腺病毒包装简要流程
1)构建表达 siRNA/miRNA 的腺病毒载体
2)采用 PacI 消化纯化的质粒。
3)消化好的腺病毒表达载体转染 293A 细胞,收获细胞以制备病毒粗提液。
4)将病毒粗提液感染 293A 细胞以扩增病毒。
5)分装,-80℃保存。
1.3、慢病毒和腺病毒的比较
慢病毒载体系统和腺病毒载体系统比较
| 病毒表达系统 | 慢病毒表达系统(Lentivirus) | 腺病毒表达系统(Adenovirus) |
| 病毒基因组 | RNA病毒 | 双链DNA病毒 |
| 复制 | 自主复制 | 自主复制 |
| 是否整合 | 病毒基因组整合于宿主基因组,长时间、稳定表达外源基因 | 病毒基因组游离于宿主基因组外,瞬时表达外源基因 |
| 感染细胞类型 | 感染分裂和不分裂细胞,适用于难转染的原代细胞(如神经细胞)及体内实验 | 感染分裂和不分裂细胞 |
| 表达风度 | 中水平表达 | 高水平表达 |
| 表达时间 | 慢(1-3天) | 快(1-2天) |
| 滴度 | 滴度更高可达10*8pfU/ml | 滴度更高可达10*11pfU/ml |
| 克隆容量 | 可插入不超过8kb的外源片段,滴度随插入片段长度增加而降低 | 可插入高达8kb的外源片段,滴度随插入片段长度增加而降低 |
| 免疫原性 | 低免疫原性 | 高免疫原性 |

2、构建目的基因到载体
2.1构建手段
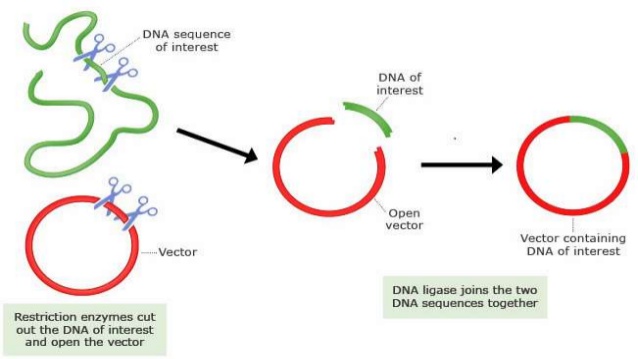
一般是根据原始质粒信息确定克隆方案,有以下两种手段。
1)如果原始质粒与载体有匹配酶切位点,采用相应的内切酶切下相应片段,回收并连接到载体,酶切,并测序鉴定
2)如果没有匹配的酶切位点,则设计带有特殊接头的引物进行PCR扩增,得到目的片段,采用相应的内切酶切下相应片段,回收并连接到穿梭载体,酶切,并测序鉴定
2.2质粒载体
2.2.1概念
能够进行自主复制的环状DNA双链结构,包括真核生物的细胞器(主要指线粒体和叶绿体)中和细菌细胞拟核区以外的环状脱氧核糖核酸(DNA)分子
2.2.2特征
质粒上常有抗生素的抗性基因,例如,四环素抗性基因或卡那霉素抗性基因等。有些质粒称为附加体(episome),这类质粒能够整合进真菌的染色体,也能从整合位置上切离下来成为游离于染色体外的DNA分子。质粒在宿主细胞体内外都可复制。通过个些特性,人们可以把一些目的DNA片断构建在质粒中,通过转化入大肠杆菌中,利用选择培养基来筛选从而不断的复制,来得到目的产物。
3、质粒DNA在大肠杆菌里转化
连接上目的基因的质粒转化大肠杆菌是为了让目的基因在大肠杆菌里扩增,然后提取质粒,以下是质粒DNA在大肠杆菌里转化的三步骤。
3.1大肠杆菌感受态细胞的制备
1)从大肠杆菌平板上挑取一个单菌落于3mlLB培养基的试管中,37℃振荡培养过夜。
2)取0.4ml菌液转接到40mlLB液体培养基中,37℃振荡培养2~3h
3)菌液转移到50ml离心管中,冰上放置10min
4)4℃离心10min(4000r/min)
5)倒出培养液,将管口倒置以便培养液流尽
6)用冰浴的0.1mol/L氯化钙10ml悬浮细胞沉淀,立即冰浴30min
7)4℃离心10min(4000r/min)
8)倒出上清液,用冰浴的0.1mol/L氯化钙2ml悬浮细胞(冰上放置)
9)分装细胞,200ul一份,4℃保存
3.2质粒DNA的转化
1)取200ul新鲜制备的感受态细胞,加入质粒DNA2ul混匀,冰浴30min
2)离心管放到42℃保温90s
3)冰浴2min
4)每管加800ulLB液体培养基,37℃培养1h(150r/min)
5)取适当体积(100ul)的复苏细胞,涂布在选择性培养基上,正置30min
6)倒置平皿37℃,12~16h,出现菌落
3.3质粒提取步骤
1)取1~4ml在LB培养基中培养过夜的菌液,12000转离心1min,弃上清
2)加250ul溶液Ⅰ/RNaseA(溶液Ⅰ为细胞悬浮液)混合液,漩涡剧烈振荡直至菌体完全重新悬浮,室温静置1-2min。
3)加入250ul溶液Ⅱ(细胞裂解液),轻柔的反复颠倒混匀5-6次。室温放置1-2min,使菌体充分裂解,直至形成澄清的裂解溶液。
4)加入350ul溶液Ⅲ(中和液),立刻轻柔地反复颠倒混匀5-6次,此时会出现白色絮状沉淀。
5)12000室温离心10min,收集上清。
6)将上清置于DNA纯化柱中,静置1-2min。
7)12000转离心1min,弃滤液。
8)加入500ul溶液PB(洗涤液)12000转离心1min,弃滤液,目的是将硅胶膜上吸附的蛋白、盐等杂质洗脱,以获得高质量质粒DNA。
9)加入500ul溶液W(去盐液),12000转离心1min,弃滤液,重复一次。
10)12000转离心3min,以彻底去除纯化柱中残留的液体。
11)将DNA纯化柱置于新的离心管中,悬浮滴加50-100ul溶液Eluent(为无菌的双蒸水,PH为8.0-8.5),室温放置2min。
12)12000转离心1min,此时管底即为高纯度的质粒DNA,质粒于-20℃保存。
质粒提取步骤:吸取液体培养基于1.5ml离心管中12000转离心1min,弃上清,吸取培养基重复离心弃上清离心,留取少量菌液作为菌种保存,可直接置于-20℃——加250ulBufferS1悬浮细菌,悬浮均匀——加250ulBufferS2温和充分的上下翻转4-6次混合均匀,使菌体裂解——加350ulBufferS3温和上下翻转12000离心10min——取上清液转移到专用的制备管(2ml)12000转离心1min,弃滤液——加500ulBufferW112000转离心1min,弃滤液——加500ulBufferW212000转离心1min,弃滤液,重复一遍——将制备管置回2ml离心管12000转离心1min——将制备管移入新的1.5ml离心管中,加60~80ulEluent或离子水,室温1min12000转离心1min——移去制备管,将有质粒的离心管于4℃或是-20℃保存
4、质粒DNA和其他包装质粒共转染293T细胞产生病毒(即病毒包装)
4.1名词解释
4.1.1293T细胞是由293细胞派生, 表达SV40大T抗原的人肾上皮细胞系, 被广泛应用于瞬时转染以过表达各种目标蛋白, 或是用以包装病毒。
4.1.2脂质体:某些细胞质中的天然脂质小体,可作为生物膜,用于捕获外源性物质后更有效地运送到靶细胞,经同细胞融合而释放。
4.2共转染的操作步骤
天:用无抗生素DMEM+10%FBS铺板293FT细胞,2ml/孔。确保第二天细胞密度达到80%-90%融合度
第二天:
1. 500ul 无血清培养基稀释2ug 表达质粒+1.5ug psPAX2+1.5ug pMD2.G
2. 500ul 无血清培养基稀释15ul 脂质体2000
3. 5min后,将DNA溶液和脂质体溶液混合,室温静止20min
4. 从6孔板中吸出1ml无血清培养基,然后滴加入1ml质粒和脂质体混合物。
5. 6-10h后,移除含有DNA-脂质体复合物的培养基,代之以正常培养液DMED+10%FB(从此刻开始算时间)。
第三天:
1.转染24h后,荧光显微镜下观察,转染效率应达到70%以上
第四天:
1.转染后48和72h分别收获含病毒的上清。
2.3000 rpm 离心20min,0.45um滤膜过滤,去除细胞沉淀。
3.12000转离心浓缩细胞、分装-80°C贮存。
4.滴度测定目的基因检定,并出具检测报告。
4.3病毒包装的原理
质粒DNA为能转录出慢病毒遗传物质(RNA),但不能翻译出慢病毒的外壳及蛋白成分的载体质粒,其同时连有目的基因和报告基因,psPAX2为能表达慢病毒外壳的质粒,其表达产物可通过粘附机制更易穿过细胞膜, pMD2.G为慢病毒的膜蛋白质粒,通过 lipofectamine2000进行三质粒共转到靶细胞基因组中,宿主基因组在表达时,随宿主基因转录出的目的基因RNA与psPAX2、pMD2.G基因翻译出的蛋白组装为慢病毒。
在上述程序中提及的“第四天”收集病毒。在第五天再用该病毒感染靶细胞,病毒进入细胞后,其遗传物质RNA反转录出DNA,该基因再整合到靶细胞的基因组中,完成转染过程,因为质粒DNA只能转录出病毒RNA和表达目的基因却不能表达出病毒的外壳和膜蛋白成分,因此其不能像普通的病毒一样在宿主细胞能反复增殖,故对宿主细胞是无害的并且的将目的基因转然到靶细胞基因组中。
5、慢病毒感染细胞
5.1流程图
5.2感染步骤
1)铺板:将对数生长期的细胞消化重悬后,按1*105/L密度接种于12孔板,生长过夜
2)感染:将70-80%铺满12孔板中的培养液吸除,换新鲜的培养液,同时加入PBS浓度梯度稀释的病毒液,混合均匀后即可放入孵箱培养。
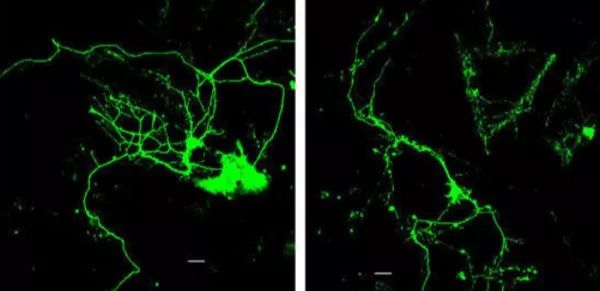
3) 24h左右可换液,48小时即可看荧光,具体根据细胞状态来看。
5.3荧光显微镜的操作流程
打开荧光器(30min内不能关闭,否则影响显微镜寿命),细胞培养板置于载物台,调节物镜和光圈,先用自然光观察视野内细胞,再关闭光,开启荧光通道,观察荧光强度,判定感染率。图片取样前可以调节曝光时间,增益值和彩色度使荧光照片最。(针对leica)
5.4注意事项内容
1.病毒浓度要适宜,太少的话细胞被感染的也少,但是病毒浓度太大,对细胞有伤害。
2.感染病毒时培养基量少,以保证病毒的浓度,在培养10h左右可根据培养基颜色加培养基。
3.在不明确细胞感染复数情况下,可进行浓度梯度感染,计算细胞感染复数。
4.在加病毒后一般24h左右可换液,48小时即可看荧光,具体时间根据细胞状态来看。
6、感染后的细胞检测方法
6.1荧光初步检测
若有荧光,则表示病毒感染成功,但并不能确定目的基因是否整合到细胞中,待进一步检测,荧光有强弱之分,与病毒加入的量有关。
6.2RNA的提取及RT-PCR检测
6.2.1原理
因为真核细胞DNA含有很多非编码区,真核生物的DNA转录成为RNA之后,经过剪切和拼接,去掉这些非编码区,才能形成真正的mRNA,,是否表达真核生物的基因并表达相应的蛋白,只能通过提取其mRNA并RT-PCR这条途径来测定
6.2.2RNA提取步骤
加1mlTrizol,吹打后移至1.5ml无菌的离心管中;加100ul氯仿剧烈振荡30s混匀,12000转15min,可看到明显分层;取上层透明液体至新的1.5ml离心管中,加等体积的异丙醇,混匀静置10min,12000转离心10min,弃上清,加1ml70%乙醇,12000转,离心10min,弃上清,风干剩余液体,最后加DEPC-水溶解RNA,电泳,粗步判定RNA纯度。
6.2.3RT-PCR步骤:
RT是一个逆转录的过程,用前提取好的总RNA,在加入引物,模版和酶,并在PCR仪的温度设置下,RNA可逆转录为cDNA。
PCR是cDNA在模版,引物,酶的作用下进行复制成双链DNA。(具体步骤省略)
6.3蛋白提取及Western检测
western-Bloting:蛋白免疫印迹(Western blotting 或 Immunoblotting)一般由凝胶电泳、样品的印迹和免疫学检测三个部分组成。步是做 SDS 聚丙烯酰胺凝胶电泳,使待测样品中的蛋白质按分子量大小在凝胶中分成带。 第二步把凝胶中已分成条带的蛋白质转移到一种固相支持物上,用得最多的材料是硝酸纤维素膜(NC 膜)和 PVDF 膜, 蛋白转移的方法多用电泳转移 (转移电泳) ,它又有半干法和湿法之分,现在大多用湿法。第三步是用特异性的抗体检测出已经印迹在膜上的所要研究的相应抗原。免疫检测的方法可以是直接的和间接的。现在多用间接免疫酶标的方法,在用特异性的抗体杂交结合后,再用酶标的第二抗体(碱性磷酸酶(AP)或辣根过氧化物酶(HRP)标记的抗抗体的抗体)杂交结合,再加酶的底物显色或者通过膜上的颜色或 X 光底片上暴光的条带来显示抗原的存在。该技术被广泛应用于蛋白表达水平的检测中。
 关注我们
关注我们